We explore the capabilities of the
Single Chain Mean Field Theory (SCMFT) developed at the URV in direct applications to the design and conception of drug carriers. The method is very competitive when compared to Monte Carlo simulations and is therefore suited for an exhaustive exploration of the compositions and molecular structures, such as those required to properly tune the properties of drug carriers. This computational method best suits the purposes of this area, since it can describe such nanoscale objects including details at the molecular level. In addition, we hope to fill with this comprehensive method the huge lack of even qualitative modeling in the domain of drug carriers.
The method exactly accounts for the configurations of a single microscopically detailed chain at the molecular level. The interactions between different chains are described self-consistently through a mean molecular field. This mean field is found self-consistently. This method can be applied to solutions of linear or branched polymers, solutions of low-molecular weight surfactants and various additives, and mixtures of various components. It can be extended for the description of ordered phases, like liquid crystals, and to disordered structures as gels. The methodology of the SCMFT by its construction can be especially successful when used in parallel for the description of nano-objects like polymeric drug carriers.(Read more)

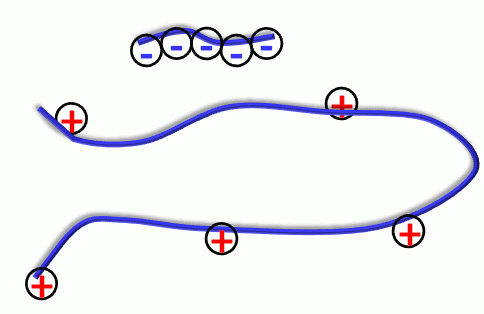 s on the cell membranes. Usually the drug carrier designed for the delivery of a charged polymer is composed of a diblock copolymer: one block is charged and forms a complex with the drug while the other block is neutral and is devised to form the corona around the aggregate, aiming at shielding the complex from the solution.
s on the cell membranes. Usually the drug carrier designed for the delivery of a charged polymer is composed of a diblock copolymer: one block is charged and forms a complex with the drug while the other block is neutral and is devised to form the corona around the aggregate, aiming at shielding the complex from the solution.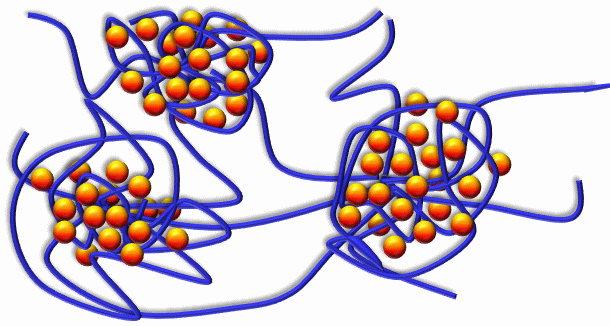 The most promising drug delivery systems are based on degradable physical gels. The target is the control of the gel architecture: structure and size of crosslinks, the distances between the hydrophobic nuclei that will store the drug, the strength of the drug binding, the drug partition coefficient and diffusion coefficients. The control of these variables will allow for a fine tuning and adjustment of the release rate and drug density profile inside the gel for a given particular need. Such modeling problems are not yet solved except in several empirical models, only suitable for one particular system. Any qualitative knowledge as well as any modeling tool will have a high impact in this domain. These gels are designed for prolonged delivery of drugs and there is a strong industrial need for a further development of this theoretical method. The system of particular interest is hydrogels PEO-PLA-PEO, where the cores of the gels are formed of Poly-Lactic acid, degrading under physiological conditions to metabolites.
The most promising drug delivery systems are based on degradable physical gels. The target is the control of the gel architecture: structure and size of crosslinks, the distances between the hydrophobic nuclei that will store the drug, the strength of the drug binding, the drug partition coefficient and diffusion coefficients. The control of these variables will allow for a fine tuning and adjustment of the release rate and drug density profile inside the gel for a given particular need. Such modeling problems are not yet solved except in several empirical models, only suitable for one particular system. Any qualitative knowledge as well as any modeling tool will have a high impact in this domain. These gels are designed for prolonged delivery of drugs and there is a strong industrial need for a further development of this theoretical method. The system of particular interest is hydrogels PEO-PLA-PEO, where the cores of the gels are formed of Poly-Lactic acid, degrading under physiological conditions to metabolites.
